.svg)
Noteworthy Datasets on Neurodegenerative Diseases
Shraddha Dumawat, Deepthi Das
%20(1).svg)
September 8, 2022

Neurodegenerative diseases (ND) are very complex disorders. Though there has been significant progress in the understanding of neurodegenerative diseases, more focus is needed on developing early diagnostic tools, providing access to more effective personalized therapies, and understanding more about misfolded proteins and their role in ND. Advanced technologies such as robotic microscopy, CRISPR screening, machine-learning methods, and high-throughput screening have increased our ability to capture data in ND research multifold. However, this mass accumulation of data has not yet translated into diagnostic and therapeutic solutions to effectively treat ND and needs to be tapped to generate actionable insights. Here, we have curated 10 datasets, each of which has made a relevant contribution to further the understanding of important ND such as amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), Huntington's disease (HD), and Alzheimer's disease (AD).
Explore these interesting datasets about the human single-nuclei transcriptomic atlas for substantia nigra, differential gene expression in ND patients versus healthy controls, the epigenetic landscape of normal aging in Alzheimer’s disease, gender-based differences in age-related ND, etc. You can find more highly curated datasets on ND (see figure below) from different repositories that can be visualized and analyzed using our DataOps platform, Polly.

Altered expression of histamine signaling genes in autism spectrum disorder
Dataset ID: GSE102741_GPL11154
Year of Publication: 2017
Total Samples: 52
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication, Raw data
The histaminergic system (HS) is critical in cognition, sleep, and other behaviors. Although not well studied in autism spectrum disorder (ASD), HS is implicated in many neurological disorders, some of which share comorbidity with ASD. Preliminary studies suggest that antagonism of histamine receptors 1-3 reduces symptoms and specific behaviors in ASD patients and relevant animal models. In addition, the HS mediates neuroinflammation, which may be heightened in ASD. The authors have used RNA sequencing (RNA-seq), investigated the genome-wide expression, as well as a focused gene set analysis of key HS genes (HDC, HNMT, HRH1, HRH2, HRH3, and HRH4) in postmortem dorsolateral prefrontal cortex (DLPFC) initially in 13 subjects with ASD and 39 matched controls.

The authors noticed that while there was no significant diagnosis effect on any of the individual HS genes, expression of the gene set of HNMT, HRH1, HRH2, and HRH3 was significantly altered. Curated HS gene sets were also significantly differentially expressed. This study represents the first specific analysis of the expression of histamine-related genes in ASD and suggests that these genes may collectively be dysregulated. Understanding the physiological relevance of an altered HS may suggest new therapeutic options for treating ASD.
Distinct brain transcriptome profiles in c9orf72-associated and sporadic ALS
Dataset ID: GSE67196_GPL11154
Year of Publication: 2015
Total Samples: 53
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication, Raw data
To assess whether aberrant expression of repetitive element sequences is observed in amyotrophic lateral sclerosis (ALS), the authors analyzed RNA sequencing data from C9orf72-positive and sporadic ALS cases, as well as healthy controls. Transcripts from multiple classes and subclasses of repetitive elements (Long interspersed nuclear elements (LINEs), endogenous retroviruses, DNA transposons, simple repeats, etc.) were significantly increased in the frontal cortex of C9orf72 ALS patients. A large collection of patient samples representing both C9orf72 positive and negative ALS, ALS/frontotemporal lobar degeneration (FTLD), and FTLD cases were used to validate the levels of several repetitive element transcripts.
These analyses confirmed that repetitive element expression was significantly increased in C9orf72-positive compared to C9orf72-negative or control cases. While previous studies suggest an important link between TDP-43 and repetitive element biology, this data indicates that TDP-43 pathology alone is insufficient to account for the observed changes in repetitive elements in ALS/FTLD. Instead, it was found that repetitive element expression positively correlated with RNA polymerase II activity in the postmortem brain. Also, pharmacologic modulation of RNA polymerase II activity altered repetitive element expression in vitro. This leads to the conclusion that increased RNA polymerase II activity in ALS/FTLD may lead to increased repetitive element transcript expression, a novel pathological feature of ALS/FTLD.

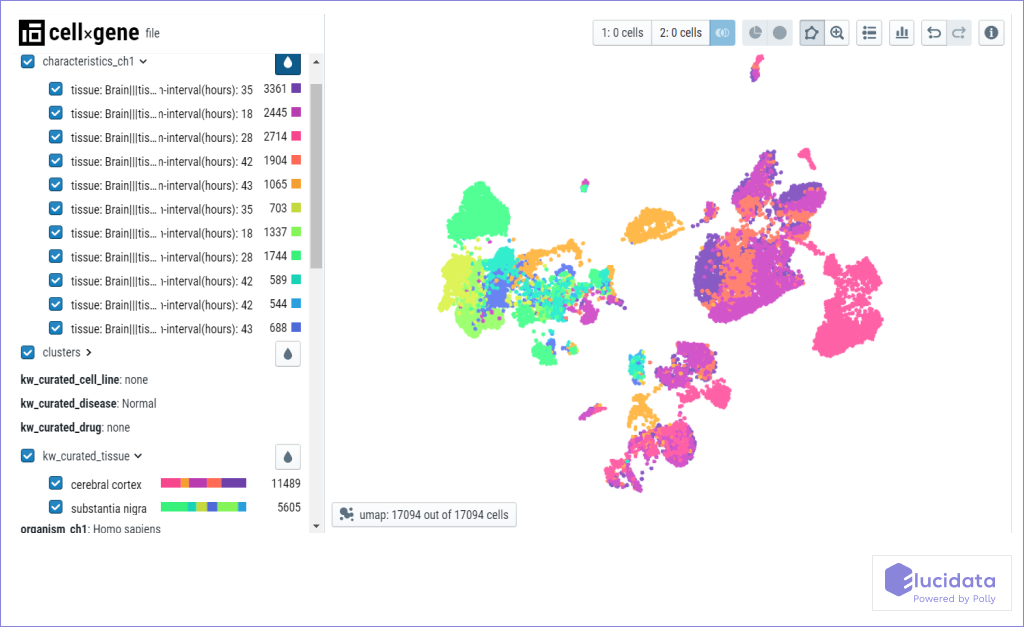
A human single-cell atlas of the substantia nigra reveals novel cell-specific pathways associated with the genetic risk of Parkinson’s disease and neuropsychiatric disorders.
Dataset ID: GSE140231_GPL20301
Year of Publication: 2020
Total cells: 17094
Experiment type: Single cell Transcriptomics
Organism: Homo sapiens
Reference link: Publication, Raw data
The authors describe the first human single-nuclei transcriptomic atlas for substantia nigra (SN), generated by sequencing ~ 17,000 nuclei from matched cortical and SN samples. They show that common genetic risk for Parkinson’s disease (PD) is associated with dopaminergic neuron (DaN)-specific gene expression including mitochondrial functioning, protein folding, and ubiquitination pathways. They have also identified a distinct cell-type association between PD risk and oligodendrocyte-specific expression implicating metabolic and gene expression regulation networks. Beyond PD, we find SN DaNs and GABAergic neurons to be associated with different neuropsychiatric disorders, particularly schizophrenia (SCZ) and bipolar disorder (BP). They have identified distinct cortex/SN associations with SCZ genetic risk for both excitatory (synaptic functioning) and dopaminergic neurons (mitochondrial functioning and synaptic signaling). Conditional analyses show that independent sets of loci associate distinct neuropsychiatric disorders with the same neuronal types. This atlas guides our aetiological understanding by associating SN cell-type expression profiles with specific disease risks.

mRNA-Seq expression profiling of human post-mortem BA9 brain tissue for Huntington's Disease (HD) and neurologically normal individuals
Dataset ID: GSE64810_GPL11154
Year of Publication: 2015
Total Samples: 69
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication, Raw data
Huntington's Disease (HD) is a devastating neurodegenerative disorder that is caused by an expanded CAG trinucleotide repeat in the Huntingtin (HTT) gene. The authors present a genome-wide analysis of mRNA expression in the human prefrontal cortex from 20 HD and 49 neuropathologically normal controls using next-generation high-throughput sequencing. Surprisingly, 19% (5,480) of the 28,087 confidently detected genes are differentially expressed (FDR<0.05) and are predominantly up-regulated. A novel hypothesis-free geneset enrichment method that dissects large gene lists into functionally and transcriptionally related groups discovers that the differentially expressed genes are enriched for immune response, neuroinflammation, and developmental genes. Markers for all major brain cell types are observed, suggesting that HD invokes a systemic response in the brain area studied. Unexpectedly, the most strongly differentially expressed genes are a homeotic gene set (represented by Hox and other homeobox genes), that are almost exclusively expressed in HD, a profile not widely implicated in HD pathogenesis. The role of inflammation and the significance of non-neuronal involvement in HD pathogenesis suggest anti-inflammatory therapeutics may offer important opportunities in treating HD.


Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease
Dataset ID: GSE104704_GPL18573
Year of Publication: 2018
Total Samples: 62
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication, Raw data
Aging is the strongest risk factor for AD, although the underlying mechanisms remain unclear. The chromatin state, in particular through the mark H4K16ac, has been implicated in aging and thus may play a pivotal role in age-associated neurodegeneration. Here the authors compared the genome-wide enrichment of H4K16ac in the lateral temporal lobe of AD individuals against both younger and elderly cognitively normal controls.
They find that while normal aging leads to H4K16ac enrichment, AD entails dramatic losses of H4K16ac in the proximity of genes linked to aging and AD. Their analysis highlights the presence of three classes of AD-related changes having distinctive functional roles. Furthermore, they discovered an association between the genomic locations of significant H4K16ac changes with genetic variants (SNPs) identified in prior AD genome-wide association studies (GWAS) and with expression quantitative trait loci (eQTLs). Their results establish the basis for an epigenetic link between aging and AD.

Gene expression changes in the course of normal brain aging are sexually dimorphic.
Dataset ID: GSE11882_GPL570
Year of Publication: 2008
Total Samples: 173
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication, Raw data
Summary:
Gene expression profiles were assessed in the hippocampus, entorhinal cortex, superior-frontal gyrus, and postcentral gyrus across the lifespan of 55 cognitively intact individuals aged 20-99 years. Perspectives on global gene changes that are associated with brain aging emerged, revealing two overarching concepts.
First, different forebrain regions exhibited substantially different gene profile changes with age. For example, comparing equally powered groups, 5,029 probe sets were significantly altered with age in the superior-frontal gyrus, compared with 1,110 in the entorhinal cortex. Prominent change occurred in the sixth to seventh decades across cortical regions, suggesting that this period is a critical transition point in brain aging, particularly in males.
Second, clear gender differences in brain aging were evident, suggesting that the brain undergoes sexually dimorphic changes in gene expression not only in development but also in later life. Globally across all brain regions, males showed more gene change than females. Further, gene ontology analysis revealed that different categories of genes were predominantly affected in males vs. females.
These data open opportunities to explore age-dependent changes in gene expression that set the balance between neurodegeneration and compensatory mechanisms in the brain and suggest that this balance is set differently in males and females, an intriguing idea.

Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer's disease.
Dataset ID: GSE134576_GPL21697
Year of Publication: 2019
Total cells: 6969
Experiment type: Single cell Transcriptomics
Organism: Homo sapiens
Reference link: Publication, Raw data
Summary:
In this study, the authors performed mass cytometry of peripheral blood mononuclear cells and detected an immunologic signature of AD characterized by increased numbers of CD8+ T effector memory CD45RA+ (TEMRA) cells. CD8+ TEMRA cells were negatively associated with cognition and single cell RNA sequencing revealed their cytotoxic effector function. Strikingly, they discovered identical, shared T cell receptors (TCRs) of clonally expanded CD8+ TEMRA cells in cerebrospinal fluid (CSF) of three AD patients. Deep TCR sequencing, machine learning, and peptide screens identified the HLA-B*08:01-restricted Epstein-Barr virus trans-activator protein BZLF1 as the cognate antigen of a novel AD CSF TCR . These results provide the first evidence of clonal, antigen-specific T cells patrolling the intrathecal space of brains affected by age-related neurodegeneration.

Molecular signatures underlying neurofibrillary tangle susceptibility in Alzheimer’s disease
Dataset ID: GSE129308_GPL24676_NFT
Year of Publication: 2020
Total cells: 37931
Experiment type: Single cell Transcriptomics
Organism: Homo sapiens
Reference link: Publication, Raw data
Summary:
Tau aggregation in neurofibrillary tangles (NFTs) is closely associated with neurodegeneration and cognitive decline in Alzheimer’s disease (AD). However, the molecular signatures that distinguish between aggregation-prone and aggregation-resistant cell states are unknown. In this study, the authors developed methods for the high-throughput isolation and transcriptome profiling of single somas with NFTs from the human AD brain, quantified the susceptibility of 20 neocortical subtypes for NFT formation and death, and identified both shared and cell-type-specific signatures. It was found that NFT-bearing neurons shared a marked upregulation of synaptic transmission-related genes, including a core set of 63 genes enriched for synaptic vesicle cycling. Oxidative phosphorylation and mitochondrial dysfunction were highly cell-type dependent. Apoptosis was only modestly enriched, and the susceptibilities of NFT-bearing and NFT-free neurons for death were highly similar. Their analysis suggests that NFTs represent cell-type-specific responses to stress and synaptic dysfunction. They provide a resource for biomarker discovery and the investigation of tau-dependent and tau-independent mechanisms of neurodegeneration.

Metabolomics profiles of patients with Wilson disease reveal a distinct metabolic signature.
Dataset ID: ST001118_AN001817
Year of Publication: 2019
Total cells: 17094
Experiment type: Metabolomics
Organism: Homo sapiens
Reference link: Raw data
Summary:
Wilson disease is caused by a defect in a copper transporter leading to copper accumulation in the liver and brain, leading to liver and/or neuropsychiatric symptoms. This study compares the plasma metabolomics profile of patients with the genetic disorder Wilson disease, compared to healthy subjects matched by age, sex, and BMI. The authors hypothesize the acylcarnitine and primary metabolite profile will differ between patients with Wilson disease and healthy subjects and that these differences may indicate specific metabolic abnormalities.

GWAS study of Alzheimer's for APP gene
Dataset ID: icd10_G30_both_sexes_APP
Year of Publication: Ongoing project
Participants : 3352
Experiment type: GWAS
Organism: Homo sapiens
Reference link: Data
Summary:
UK Biobank is a collection of half a million individuals with paired genetic and phenotype information that has been valuable in studies of genetic etiology for common diseases and traits.
Phenotypes studied include:
To summarize, phenotypes included both data pulled from electronic medical records as well as participants' survey responses to questionnaires given online or at the clinic. The Pann-UkBioBank GWAS data consists of the multi-ancestry analysis of 7,221 phenotypes across 6 continental ancestry groups. Here, we look at the case of Alzheimer's disease.

Polly’s OmixAtlases provide FAIR biomolecular data on the Polly platform enabling researchers to carry out robust data analysis and effective consumption of omics data. Reach out to us at info@elucidata.io for more details.

%20copy%201.webp)

.webp)

.svg)
.svg)


%201%20(1).svg)







.svg)
%20(1).svg)



