.svg)
Noteworthy Datasets on Liver Diseases
Harsh Malavia, Deepthi Das
%20(1).svg)
May 25, 2022
.webp)
The ‘Monthly Dataset Roundup’ series features datasets on Polly that are of scientific value, intended to promote data sharing and reuse of multi-omics data.
This month’s roundup features datasets that capture the comprehensive molecular landscape of liver diseases. There are more than forty types of liver diseases with different molecular clues pointing toward each of them. The identification of biomarkers and other early determinants is critical as early detection can swing the balance between life and death in many cases. Here, we provide a list of curated datasets that cover datasets with important insights about major liver diseases such as liver cancers, Hepatitis, fatty liver, etc., which could accelerate these discoveries. You can find a plethora of highly curated datasets on liver diseases across repositories and different data in OmixAtlas, that can be analyzed with our DataOps platform, Polly (see figure below).
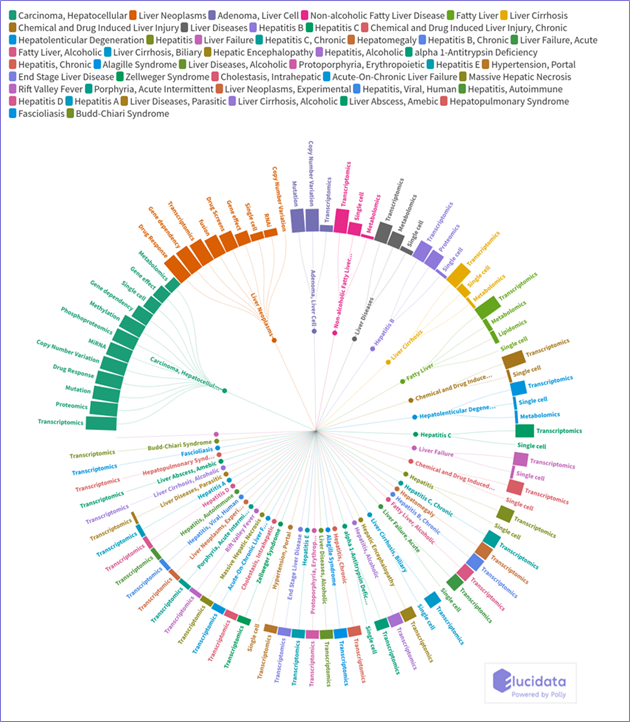
This blog is divided into five parts covering noteworthy datasets from five major types of liver diseases which are as follows:

Hepatocellular carcinoma MSK (Memorial Sloan Kettering Cancer Center) project dataset
Dataset ID: HCC_MSK*
Year of Publication: 2018
Total Samples: 268
Experiment type: CNV, mutation analysis
Organism: Homo sapiens
Reference link: Publication 1 , Publication 2
Summary:
Datasets from two cBioPortal hepatocellular carcinoma (HCC) projects submitted by the Memorial Sloan Kettering Cancer Center, New York, consisting of Copy Number Variation (CNV) and mutation data obtained by NGS capture assays. The MSK-VENTURAA study aimed to identify frequently occurring mutations in an HCC cohort compared to normal, while the MSKIMPACT study consisted of sequencing 10,000 clinical samples with advanced metastases to study their CNV profile. Datasets from both the projects can be accessed on Polly and used for downstream analysis.

TCGA_LIHC dataset
Dataset ID: LIHC_*
Year of Publication: Continuously updated
Total Samples: 2145
Experiment type: Copy Number variation, Mutation analysis, Methylation
Organism: Homo sapiens
Reference link: GDC

Summary:
The TCGA-LIHC is a large-scale project to study Liver Hepatocellular Carcinoma (LIHC) using clinical samples by correlating patient data with genotypic data obtained using NGS to better understand the genotype-phenotype relation involved in the disease.
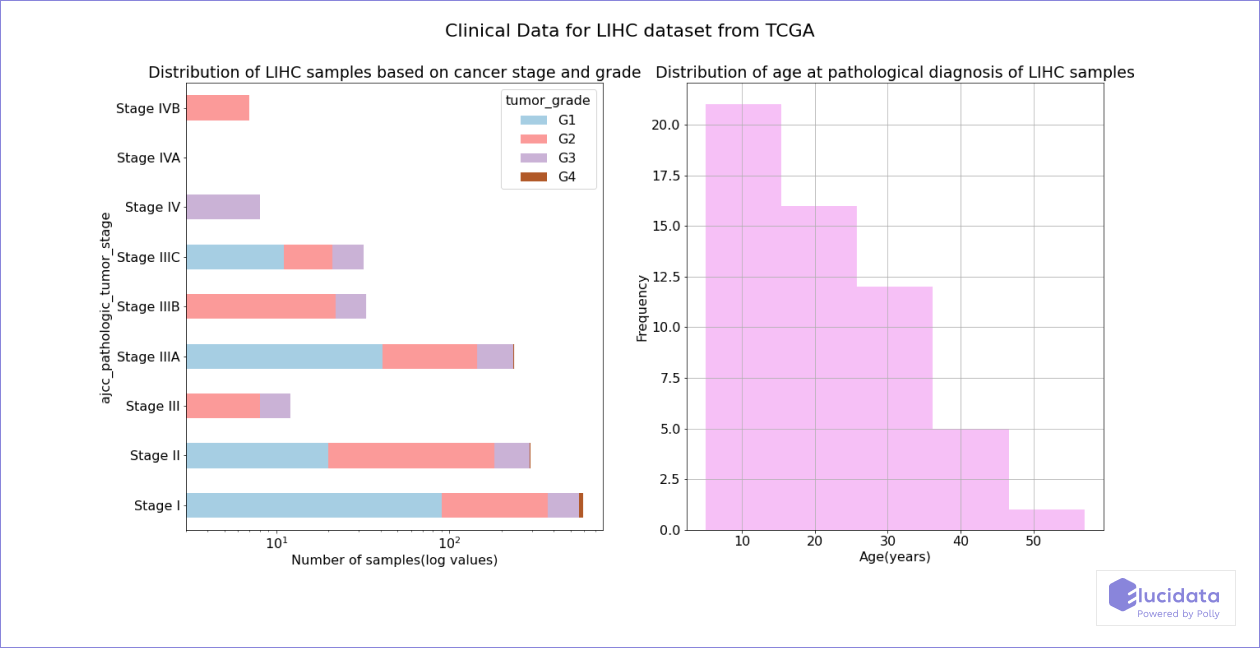
Polly-python API allows users to programmatically search and access datasets from a number of public repos using simple SQL like queries. Polly-python can also be used to access sample level metadata for datasets or projects with large number of samples like TCGA-LIHC. This can be used to quickly plot and visualize various clinical feature of the sample as demonstrated below.


Infection with Hepatitis C Virus (HCV) depends on TACSTD2, and Occludin is highly downregulated in HCC
Dataset ID: GSE69715_GPL570
Year of Publication: 2018
Total Samples: 103
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication

Summary:
Entry of HCV into hepatocytes is a complex process that involves numerous cellular factors, including the scavenger receptor class B type 1 (SR-B1), the tetraspanin CD81, and the tight junction (TJ) proteins claudin-1 (CLDN1) and occludin (OCLN).
Despite the expression of all known HCV-entry factors, in-vitro models based on hepatoma cell lines do not fully reproduce the in-vivo susceptibility of liver cells to primary HCV isolates, implying the existence of additional host factors which are critical for HCV entry and/or replication.
By performing transcriptomic analyses of tumorous and non-tumorous liver tissue obtained from eight patients with HCV-associated hepatocellular carcinoma, the researchers identified TACSTD2 as a novel regulator of two major HCV entry factors, CLDN1 and OCLN, which are strongly downregulated in malignant hepatocytes. These results provide new insights into the complex process of HCV entry into hepatocytes and may assist in the development of more efficient cellular systems for HCV propagation in vitro.

Transcriptomic profiling following de novo Hepatitis B vaccination reveals the role of granulocytes in non-responders
Dataset ID: GSE110480_GPL18573
Year of Publication: 2019
Total Samples: 215
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication
Summary:
As the Hepatitis B virus is wide-spread, WHO recommends vaccination from infancy to reduce acute infection and chronic carriers. However, current subunit vaccines are not 100% efficacious and leave 5-10% persistent non-responders unprotected. To handle large inter-individual variability in immune response after the first Engerix-B vaccination, the researchers employed whole blood early gene expression signatures on day 3 and 7. Immune-related pathways are differentially expressed in the responders' group mostly on day 3 and on day 7 in the non-responders. A notable difference between both groups is significant differentially expressed genes at day 0, before vaccination, showing the inter-individual variation. Further, absolute granulocyte numbers were significantly higher in non-responders.
Hence, the group concluded that there is a certain diversity in the basic innate immune system.



Long non-coding RNAs changes in the livers of NAFLD patients compared with that of healthy control
Dataset ID: GSE107231_GPL20115
Year of Publication: 2017
Total Samples: 10
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication

Summary:
Ultraconserved (uc) RNAs, a class of long non-coding RNAs (lncRNAs), are conserved across humans, mice, and rats, but the physiological significance and pathological role of ucRNAs is largely unknown. This data shows that uc.372 is upregulated in the livers of db/db mice, HFD-fed mice, and non-alcoholic fatty liver disease (NAFLD) patients. Gain-of-function and loss-of-function studies indicate that uc.372 drives hepatic lipid accumulation in mice by promoting lipogenesis. The researchers further demonstrate that uc.372 binds to pri-miR-195/pri-miR-4668 and suppresses the maturation of miR-195/miR-4668 to regulate the expression of genes related to lipid synthesis and uptake, including ACC, FAS, SCD1, and CD36.



Hepatic transcriptome signatures in patients with varying degrees of NAFLD compared to healthy normal-weight individuals
Dataset ID: GSE126848_GPL18573
Year of Publication: 2019
Total Samples: 33
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication
Summary:
NAFLD represents a spectrum of conditions ranging from simple steatosis to non-alcoholic fatty liver (NAFL), to non-alcoholic steatohepatitis (NASH) with or without fibrosis, to cirrhosis with end-stage disease. The hepatic molecular events underlying the development of NAFLD and transition to NASH are poorly understood. The above study aimed to determine hepatic transcriptome dynamics in patients with NAFL or NASH compared to healthy normal-weight and obese individuals. RNA sequencing and quantitative histomorphometry of liver fat, inflammation, and fibrosis were performed on liver biopsies obtained from healthy normal weight (n=14) and obese (n=12) individuals, NAFL (n=15) and NASH (n=16) patients. Normal weight and obese subjects showed normal liver histology and comparable gene expression profiles. Liver transcriptome signatures were largely overlapping in NAFL and NASH patients, however, clearly distinguishable from healthy normal-weight, and obese controls. Most marked pathway perturbations identified in both NAFL and NASH were associated with markers of lipid metabolism, immunomodulation, extracellular matrix remodeling, and cell cycle control.
In conclusion, the application of immunohistochemical markers of hepatocyte injury may serve as a more objective tool for distinguishing NASH from NAFL, facilitating the improved resolution of hepatic molecular changes associated with the progression of NAFLD.
Classifying distinct grades of human NAFLD employing a systems biology approach
Dataset ID: GSE46300_GPL10558
Year of Publication: 2015
Total Samples: 18
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication
Summary:
With an estimated prevalence of about 30% in western countries, NAFLD is a major public health issue. It is associated with the metabolic syndrome of insulin resistance, obesity, and glucose intolerance. Although many studies are pointing to the induction of insulin resistance by NAFLD, causality between both phenotypes is not fully clarified.
This dataset investigates liver samples from patients with varying severities of steatosis in an integrative approach employing transcriptomics, serum biomarker profiling, metabolomics data, and systems biology models.




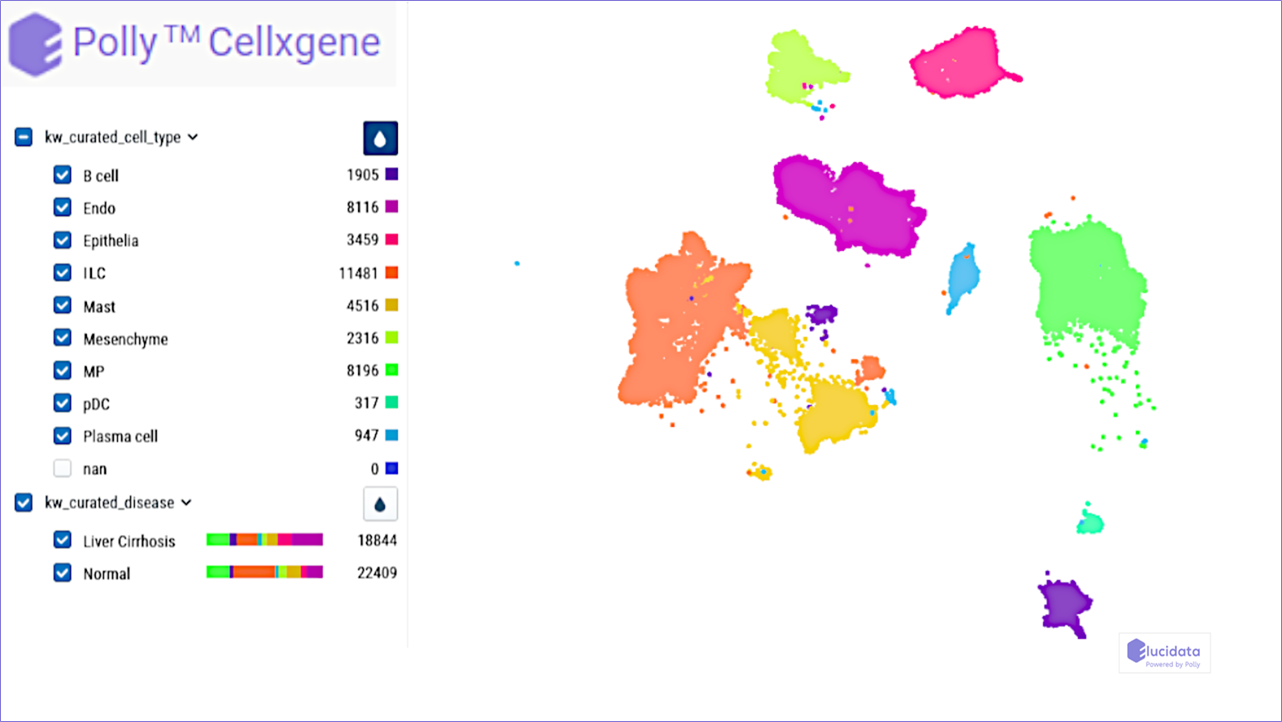
Resolving the fibrotic niche of human liver cirrhosis using single-cell transcriptomics
Dataset ID: GSE136103_GPL20301
Year of Publication: 2019
Total Samples: 24 samples (91240 cells)
Experiment type: scRNA-Seq
Organism: Homo sapiens
Reference link: Publication

Summary:
Liver cirrhosis is a major cause of death worldwide and is characterized by extensive fibrosis. There are currently no effective antifibrotic therapies available. To obtain a better understanding of the cellular and molecular mechanisms involved in disease pathogenesis and enable the discovery of therapeutic targets, this dataset profiles the transcriptomes of more than 100,000 single human cells, yielding molecular definitions for non-parenchymal cell types that are found in healthy and cirrhotic human liver.
This work dissects unanticipated aspects of the cellular and molecular basis of human organ fibrosis at a single-cell level, and provides a conceptual framework for the discovery of rational therapeutic targets in liver cirrhosis.

Transcriptome analysis of fetal and adult liver samples
Dataset ID: GSE61276_GPL10558
Year of Publication: 2014
Total Samples: 103
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication
Summary:
The study includes 106 individuals, 14 fetal and 92 adult samples, no replicates. Liver samples from 14 fetuses were obtained at gestational week 8-12. Adult liver samples were collected from 50 organ donors who had met accidental death and 42 liver samples from patients undergoing liver resection due to malignant tumors, most commonly from patients with metastatic colon cancers. Liver biopsies from these patients were collected from 'healthy' tissue that showed no visible pathological changes compared to the adjacent tumor.

Large-scale screening of circulating microRNAs in individuals with HIV-1 mono-infection reveals specific liver damage signatures
Dataset ID: GSE141522_GPL16791
Year of Publication: 2019
Total Samples: 91
Experiment type: Transcriptomics
Organism: Homo sapiens
Reference link: Publication

Summary:
Human immunodeficiency virus type 1 (HIV-1)-induced inflammation and/or long-term antiretroviral drug toxicity may contribute to the evolution of the liver disease. We investigated circulating plasma microRNAs (miRNAs) as potential biomarkers of liver injury in patients mono-infected with HIV-1.
The researchers performed large-scale deep sequencing analyses of small RNA levels on plasma samples from patients with HIV-1 mono-infection that had elevated or normal levels of alanine aminotransferase (ALT) or focal nodular hyperplasia (FNH). Hepatitis C virus (HCV) mono-infected patients were also studied. Compared to healthy donors, patients with HIV-1 or HCV mono-infection showed significantly altered levels of 25 and 70 miRNAs, respectively.
MiR-122-3p and miR-193b-5p were highly up-regulated HIV-1 mono-infected patients with elevated ALT or FNH, but not in HIV-1 patients with normal levels of ALT. These results reveal that HIV-1 infections impacted liver-related miRNA levels in the absence of an HCV co-infection, which highlights the potential of miRNAs as biomarkers for the progression of liver injury in HIV-1 infected patients.



Polly’s OmixAtlases provide FAIR biomolecular data on the Polly platform enabling researchers to carry out robust data analysis and effective consumption of omics data. Reach out to us at info@elucidata.io for more details.

%20copy%201.webp)

.webp)

.svg)
.svg)


%201%20(1).svg)







.svg)
%20(1).svg)



